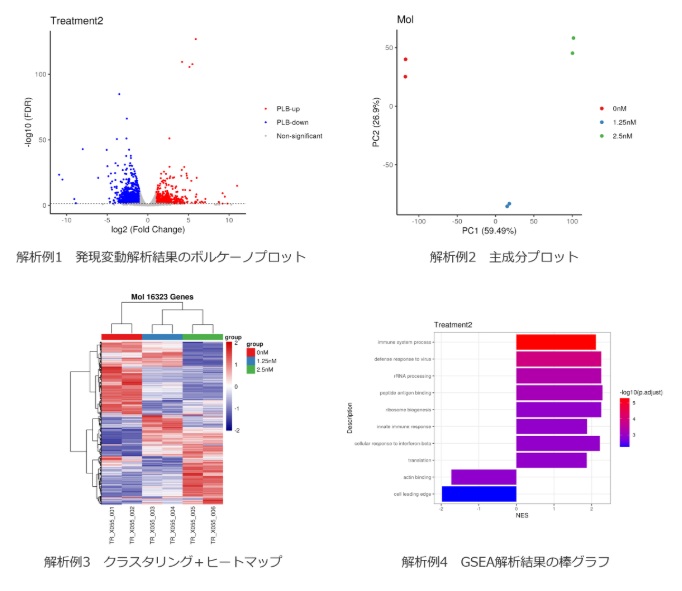
次世代シーケンサーを用いて遺伝子発現変動を網羅的に解析します。
| No | ライブラリー作製キット | RNA精製 方法 |
FFPE由来 RNA対応 |
特徴 |
|---|---|---|---|---|
| 1 | TruSeq Stranded mRNA Library Prep(*1) | poly A RNA精製 | × | ・最もスタンダードな手法。 ・分解の見られるRNAは適さない。 |
| 2 | TruSeq RNA Exome(*2) | - | 〇 | ・ヒト遺伝子(21,415)のコーディング領域のみを濃縮して解析。 ・分解の見られるRNAでも解析可能。 |
| 3 | TruSeq Stranded Total RNA Library Prep | rRNA除去 | × | ・polyAを持たないタイプのnon-coding RNAもシーケンス可能。 ・分解の見られるRNAでも解析可能。 |
| 4 | SureSelect XT RNA direct, SureSelect Mouse All Exon(*3) | - | 〇 | ・マウス遺伝子のコーディング領域のみを濃縮して解析。 ・分解の見られるRNAでも解析可能。 |
| ライブラリ作製キット | TruSeq Stranded mRNA Library Prep |
|---|---|
| 機種 | NovaSeq 6000 |
| 平均データ量 / サンプル | 約40Mリード以上 |
| シーケンス方法 | Paired End / Multiplex |
| バイオインフォマティクス解析 | スプライシングパターン予測、正規化された発現レベルの算出 発現レベルのサンプル間比較、GO解析 |
| 納期 | 品質評価通過後、約2.5ヶ月(*) |
* 同時に解析を行うサンプル数が多い場合には、別途お打ち合わせの上決定いたします。
● ヒト、マウス、ラット以外の生物種の解析をご希望の場合は別途ご相談ください。
| ライブラリ作製キット | TruSeq Stranded mRNA Library Prep、 TruSeq Stranded Total RNA Library Prep |
TruSeq RNA Exome SureSelect XT RNA direct + SureSelect Mouse All Exon |
サンプルの種類 | 精製Total RNA | 精製Total RNA |
|---|---|---|
| RNA量 | 3μg | 0.5μg |
| 濃度(*1) | 65ng/μL以上 | 20ng/μL以上 |
| RIN値(*2) | 7以上 | -(*3) |
*1 サンプルの定量はAgilent 2100バイオアナライザまたはAgilent 2200 TapeStationを用いた方法を推奨しております。
*2 サンプルの(バイオアナライザによる)電気泳動図がお手元にある場合にはご提供をお願いします。
*3 200nt以上のRNA断片が50%以上(DV200≥50%)の品質を推奨しています。
お問い合わせフォーム よりお問い合わせください。
| サービス項目 | 価格(税抜) | 納期 |
|---|---|---|
| Transcriptome Sequencing | お問い合わせ | お問い合わせ |
/
※お問い合わせは完了しておりません。